1 临床资料
患者,男性,67岁,右利手。因“行走不稳1年,记忆力下降半年,加重3月”于2022年9月8日入住山东第一医科大学附属省立医院神经内科认知障碍病房。
1.1 现病史
患者1年前无明显诱因出现走路不稳,表现为直线行走困难,仍可自行行走,伴性格改变,表现为脾气暴躁、易怒。半年前症状逐渐加重,出现记忆力减退,以近记忆力下降为主,表现为忘记刚放置物品的位置,忘记刚发生的事;伴有反应迟钝,说话、动作较前迟缓;言语较前欠流利。偶有饮水呛咳,夜尿增多,偶有夜间睡眠中大喊大叫。3月前症状逐渐加重,于外院行颅脑MRI示双侧基底节、丘脑、额叶及大脑半球皮层多发异常信号,予美多芭及营养神经等治疗,症状未见明显改善。患者自发病以来,睡眠欠佳,饮食差,大便正常,小便夜尿增多,体重未见明显变化。
1.2 既往史
患者50余年前有头部外伤史,遗留言语不清;“心动过速”病史。
1.3 个人史
患者吸烟30年,每天1盒,已戒烟20年;偶饮白酒,已戒酒1年;有农药、混合墙胶接触史。婚育史无特殊。
1.4 家族史
患者父母因“冠心病”去世,哥哥患有高血压、糖尿病。否认其他家族遗传疾病史。
1.5 体格检查
体温36.9 ℃,血压116/100 mmHg,心率65次/分,心、肺、腹检查未见明显异常。神经系统查体:神志清、精神可,言语欠清晰流利。床边认知查体提示近记忆力减退,计算力、定向力、理解力尚可。粗测视力正常,双瞳孔等大等圆,直径3 mm,对光反射灵敏,双眼球活动灵活。双侧鼻唇沟对称,伸舌居中,咽反射正常。四肢肌力5级、肌张力正常。双手姿势性震颤,双侧指鼻试验、双侧跟膝胫试验欠稳准,闭目难立征(+)。感觉系统检查未见异常。四肢腱反射(+),双侧Babinski征(-),Chaddock征(-)。颈软,双侧克氏征(-),布氏征(-)。
1.6 辅助检查
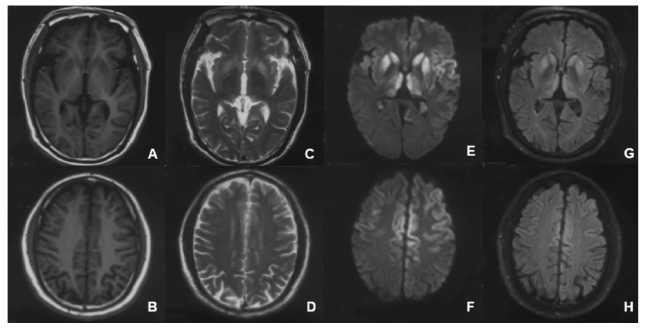
血常规、血生化、血脂、血清铜、铜蓝蛋白、甲功、叶酸、维生素B12、同型半胱氨酸、心肌酶谱、ASO、RF、抗核抗体谱定量、病毒全项、梅毒抗体、莱姆病DNA、AFP、CEA、CA19-9、PSA、尿常规、大便常规未见明显异常。肝功:总蛋白 64.5 g/L,总胆红素 34.98 μmol/L,直接胆红素 6.12 μmol/L,间接胆红素 28.86 μmol/L,尿酸 538 μmo1/L。血浆乳酸测定:2.40 mmo1/L。TORCH:风疹病毒IgG抗体(+)、巨细胞病毒IgG抗体(+)、单纯疱疹病毒IgG抗体(+)、细小病毒B19 IgG抗体(+)。完善脑脊液检查:压力为105 mmH2O,有核细胞1×106/L,蛋白0.36 g/L,葡萄糖 2.99 mmol/L,氯 127.2 mmol/L,脱落细胞可见少量成熟淋巴细胞;单纯疱疹病毒、EB病毒、巨细胞病毒DNA未见异常。自身免疫性脑炎抗体谱(江苏先声医学诊断),包括 NMDAR、LGI1、CASPR2、GABABR、AMPAR1、AMPAR2示阴性。脑脊液14-3-3蛋白(中国疾病预防控制中心)示阳性。血PRNP基因序列(中国疾病预防控制中心)未发现突变,129位氨基酸多态性为M/V型,219位氨基酸多态性为E/E型。神经心理学检查:小学学历,MMSE 24分,MoCA 17分,NPI 4分。视频脑电图示发作间期脑电图,未见明显异常。颅脑MR平扫和增强:双侧基底节、丘脑、额顶叶皮层多发对称性异常信号,代谢性脑病或克雅氏病待排,请结合临床进一步检查。(见图1)
图1 患者颅脑MRI平扫A、B)T1轴位,C、D)T2轴位,E、F)DWI轴位,G、H)FLAIR 轴位,示双侧基底节、丘脑、额顶叶皮层多发对称性异常信号。Fig.1 Patient Brain MRI scan A、B) T1WI axis,C、D)T2WI axis,E、F)DWI axis,G、H)FLAIR axis,Multiple asymmetric signals in bilateral basal ganglia, thalamus and frontal parietal cortex |
Full size|PPT slide
1.7 诊疗经过
诊断为临床很可能的克雅病。对症治疗:给予改善认知、营养神经治疗。由于患者长期在外地居住,患者出院后电话随访,家属诉行走不稳症状逐渐加重,无法独立行走,认知功能障碍逐渐进展,严重影响日常生活,伴言语表达减少,说话费力,构音不清。出院1月后于我院门诊就诊,复查MMSE 13分,脑电图轻度异常脑电图。
2 病例讨论
克雅病(Creutzfeldt-Jakob disease,CJD)是由变异的朊蛋白引起的一种罕见的可传染的、进展性、致死性中枢神经系统变性病变,为快速进展性痴呆常见的病因之一。临床表现为快速进展性痴呆、共济失调、肌阵挛,可伴有锥体系及锥体外系症状。病理改变主要是大脑皮质神经细胞死亡、胶质细胞增生及这两者周围的组织结构发生海绵状变性。散发型CJD最常见,约占全部CJD患者的85%,发病原因多不明确,其次为家族型或医源型,变异型较为罕见。目前确诊克雅氏病的唯一方法是脑组织活检,免疫组织染色可发现脑内异常朊蛋白的沉积。由于脑组织活检创伤性较大,因此现阶段多为临床诊断。
1998年世界卫生组织提出的散发型CJD (sporadic Creutzfeldt-Jakob disease,sCJD)诊断标准,其辅助检查主要参考脑脊液14-3-3蛋白或脑电图示周期性尖-慢复合波
[1]。随着头颅MRI研究的深入,sCJD诊断标准也不断的进行修订。从2009年欧洲MRI-CJD诊断标准及2010年的美国疾病控制和预防中心推荐临床诊断标准开始更加强调MRI的诊断价值,尤其是DWI序列在sCJD早期诊断中的价值
[2]。研究发现DWI/FLAIR序列上所见异常高信号对sCJD诊断的敏感性、特异性、准确度均高
[3]。目前sCJD临床诊断根据国家疾控中心推荐的诊断标准:具有进行性痴呆,临床病程短于2年;常规检查未提示其他诊断;具备以下4种临床表现中的至少2种:(1)肌阵挛;(2)视觉或小脑障碍;(3)锥体/锥体外系功能障碍;(4)无运动型缄默症。并且以下辅助检查至少一项阳性:(1)在病程中的任何时期出现的典型的周期性尖慢复合波脑电图改变;(2)脑脊液检查14-3-3蛋白阳性;(3)MRI-DWI像或FLAIR像上存在两个以上皮质异常高信号“缎带征”和(或)尾状核/壳核异常高信号。
朊病毒蛋白PrPc由人类20.29号染色体上的PRNP基因编码,PRNP在密码子129处具有正常的遗传多态性,可能编码蛋氨酸(M)或缬氨酸(V)。当致病型PrPSc在特定条件下被蛋白酶K切割时,会产生两种片段形式:21 kD片段(1型)和19 kD片段(2型),可将其分为MM1、MM2、MV1、MV2、VV1和VV2 6种亚型
[4]。不同的基因亚型往往影响疾病表型。70%的sCJD表现出典型的快速认知功能下降伴肌阵挛的MM1/MV1型,25%的分别表现出与VV2、MV2型相关的共济失调及库鲁斑块变异伴或不伴丘脑异常信号。另外两种亚型,包括快速进行性痴呆伴肌阵挛的罕见表型MM2型及以进行性痴呆为特征的罕见表型VV1型
[5]。
不同研究发现脑脊液14-3-3蛋白对于诊断sCJD的敏感性大致相同,而特异性差别较大
[6]。2012美国神经病学学会AAN指南指出,脑脊液14-3-3蛋白检测在诊断CJD方面具有中等准确性:敏感性为92%,特异性80%,阳性似然比4.7,阴性似然比0.10
[7]。并且指出脑脊液14-3-3检测的价值将在很大程度上取决于临床医生对于患者CJD可能性的判断。同时,许多临床研究也发现14-3-3蛋白与很多神经性疾病有关,如副肿瘤性神经系统疾病、多发性硬化、桥本氏脑病等。因此,2018年Hermann等提出的sCJD的临床诊断标准中新增加了脑脊液实时震动诱导转化试验(Real-Time quaking-induced Conversion,RT-QuIC)阳性的诊断价值
[8]。RT-QuIC使用重组朊蛋白(PrP)作为底物,在体外扩增CJD患者脑脊液中的致病形式朊病毒并进行定量,RT-QuIC对sCJD的敏感性和特异性均高
[9]。
此患者综合临床表现、脑脊液14-3-3蛋白和MRI异常的演变,考虑临床很可能的CJD。PRNP序列未发现突变,排除了遗传性CJD。此患者未进行脑活检,故无法达到病理确定诊断标准。既往也曾有类似病例报道,患者开始表现为快速进展性共济失调,后期逐渐出现大脑皮质、锥体外系、锥体系症状,同时后期脑脊液及MRI出现特征性表现,最后通过脑组织活检诊断为CJD
[10-11]。此患者在后期随访中需要再完善脑脊液RT-QuIC检查。
3 小结
CJD临床罕见,临床表现复杂,尤其是在疾病早期不易识别。随着病情进展,逐渐出现快速进展的痴呆。CJD目前没有有效的治疗方法,主要以对症治疗为主,病程多不超过2年,预后极差。


{kind=link}