WS 1型是与位于4号染色体WFS1基因相关的常染色体隐性遗传的神经变性疾病。在临床上较为罕见,发病率为1/68000 ~ 1/770000
[1-2]。其主要临床特征包括尿崩症(DI)、糖尿病(DM)、视神经萎缩(OA) 和耳聋(D),因此又被称为DIDMOAD综合征。青少年起病的胰岛素依赖性糖尿病和视神经萎缩是典型WS诊断所必需的。WS的临床表现有高度异质性,在同一家庭同一位点的变异可能出现不同的表型,可以仅出现典型临床特征中的部分表现,称为不完全WS或类WS
[3]。研究发现WFS1基因除隐性遗传模式,还存在部分杂合位点表现出显性遗传模式,其临床表型与经典WS不尽一致
[4-5],也被称为WFS1样综合征。WS预后不佳,死亡年龄中位数为30岁(25~49岁)
[1]。神经系统表现主要包括小脑性共济失调、周围神经病、癫痫、痴呆等
[6],患者最终可因脑干萎缩导致中枢性呼吸衰竭死亡
[1]。临床表现为颞叶、岛叶等部位受累为主的WS,目前罕有报道。本文对一例首诊为自身免疫性脑炎的不完全WS患者的临床资料进行整理,汇报如下。
1 病例资料
1.1 病史
患者吴某,男性,42岁,中专文化。因头痛、言语困难伴记忆力下降5个月余于2020年7月收入我院认知障碍病房。
患者于2020年2月9日无明显诱因出现头痛,持续性额顶部搏动性疼痛,头痛半小时前有视物模糊,无发热,无恶心呕吐。3天后患者出现言语困难,不能交流,当地医院行颅脑MRI检查提示左侧颞叶、岛叶、顶叶DWI高信号,MRA未见异常,发泡实验为阴性,颅脑MRV未见明显异常。脑脊液(CSF)细胞数3*106/L,蛋白0.56g/L,考虑自身免疫性脑炎可能。2月21日转至当地省级医院,以“头痛13天,言语混乱13天”收入院。入院后颅脑MRI示左侧颞、岛、顶、枕叶及海马区、右侧额叶异常信号,脑炎可能。颅脑SWI未见明显异常,心脏超声:左室心肌致密化不全,左心扩大,室间隔增厚,主动脉瓣、二尖瓣轻中度反流,三尖瓣、肺动脉瓣轻度反流。脑电图提示高度异常,广泛性低高波幅复形慢波,并有阵发性波幅增高。动态心电图提示窦性心动过缓,偶发房性早搏,偶发室性早搏,有时呈间位,P-R间期略延长,左室高电压,ST-T改变。电测听检查提示全频重度耳聋。血常规、血氨、皮质醇、甲状腺功能 、肿瘤标志物、结核感染T细胞、乙肝五项、血及脑脊液乳酸、梅毒二项及自身免疫性脑炎系列均无异常。脑脊液细胞数2*106/L,蛋白0.65g/L,葡萄糖3.29 mmol/L,氯126 mmol/L,3月1日复查颅脑MRI示左侧颞顶叶皮层异常信号,结合病史,考虑炎症或遗传代谢性疾病,予以外送神经系统遗传代谢病基因谱检测。住院期间予以地塞米松(每天静滴10 mg,共4天)以及更昔洛韦、拉莫三嗪、艾地苯醌等药物治疗,患者病情稳定后出院。患者出院后仍有发作性头痛,言语困难及记忆力下降,为进一步诊治来我院就诊。
1.2 既往史
双眼高度近视30余年;听力下降、先兆偏头痛病史20余年;糖尿病病史3年,平时口服二甲双胍及阿卡波糖,未监测血糖。
1.3 家族史
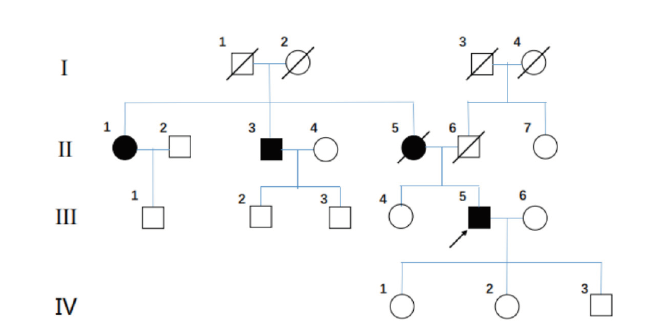
母亲(Ⅱ:5)患有糖尿病、白内障及心力衰竭(起病年龄不详,60岁因心力衰竭去世)。舅舅(Ⅱ:3)有白内障、糖尿病及冠脉支架植入术病史。姨妈(Ⅱ:1)患有白内障。舅舅家一位表弟(Ⅲ:2)有糖尿病及近视,另外一位表弟(Ⅲ:3)有近视。(详见图1)
图1 家系图Fig 1 Family tree Note:II-1: 60 years old, carried by the Wolfram gene c.975C>A mutation, with cataracts; II-2: 67 years old, with a mutation in the Wolfram gene c.975C>A, with a history of diabetes, cataract and coronary stenting; II-5: Died at the age of 67, suffering from diabetes, cataracts and heart failure; III-2: 43 years old, with diabetes mellitus and high myopia; III-3: 40 years old, myopia; IV.-1: 18 years old, with myopia. |
Full size|PPT slide
1.4 体格检查
体温 36.5℃,脉搏57次/分,呼吸18次/分,血压120/80 mmHg,神清,言语欠流利,双侧瞳孔等大等圆,直径约3.0 mm,对光反射灵敏。双眼各方向活动自如,无眼震。面纹对称,伸舌居中。颈软,四肢肌力正常。双侧痛温觉对称存在。双侧腱反射对称存在。双侧巴氏征阴性。指鼻试验及跟膝胫试验稳准,闭目难立征阴性。心脏、肺、腹部体格检查均未见明显异常,双下肢无水肿。
1.5 认知测试
MMSE29分(正常值>24分),Moca21分(正常值>24分)。Rey-O图形测验36分(正常值>32分),Rey-O图形回忆15.5分(正常值>9分)。波士顿命名测验15/30(正常值>20分)。听觉词语学习测验:N1 5/12,N2 7/12,N3 8/12,N4 3/12,N5 3/12(正常值:N4>4分,N1+N2+N3+N4+N5>20分)。数字广度:顺背3+倒背2=5(顺背正常值5-8)。连线测验2总时间133秒(正常值<200)。符号数字转换测验90秒正确填写38个(正常值>29)。
1.6 实验室检查
尿常规提示微量蛋白,糖化血红蛋白:8.2%,血常规、大便常规+潜血、肝肾功、电解质、同型半胱氨酸、肿瘤标志物、ENA谱、抗心磷脂抗体、抗肾小球基底膜抗体、甲状腺功能、HIV抗体、RPR及TPPA、血乳酸、血叶酸及维生素B12水平、心肌标志物、转铁蛋白、免疫球蛋白、高敏C反应蛋白、铜蓝蛋白、β2微球蛋白、C肽+胰岛素水平均正常。
1.7 影像学检查
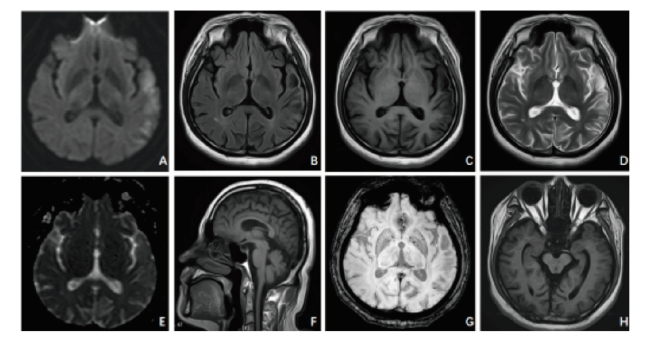
胸部、腹部及泌尿系CT未见明显异常。肌电图未见明显异常,脑电图示双侧大量θ波和一些δ波,有时δ波阵发出现。颅脑MRI考虑左侧颞叶新近梗死灶可能,双侧额顶叶及侧脑室旁多发缺血灶,脑萎缩(图2 A-F)。SWI未见明显异常(图2G)。头颅ASL提示左侧颞枕叶脑组织血流灌注减低。眼眶MR提示双侧眼球前后径增大,双侧视神经未见明显变细或增粗(图2H)。眼科会诊:OCT检查提示神经纤维层厚度和黄斑GCC厚度局限变薄,视野向心性缩小,高度近视,未见视网膜色素变性改变。
图2 颅脑和视神经磁共振影像图Figure 2 Magnetic resonance imaging of brain and optic nerves Note:A: Axial diffusion-weighted imaging (DWI) reveals high signal intensity in the left temporal lobe; B: Axial fluid-attenuated inversion recovery (FLAIR) image; C: Axial T1-weighted image;D: Axial T2-weighted image;E: Axial apparent diffusion coefficient (ADC) map; F: Sagittal T1-weighted image;G: Susceptibility-weighted imaging (SWI) of the brain shows no evidence of microhemorrhages (axial);H: Orbital MRI reveals no optic nerve atrophy (axial). |
Full size|PPT slide
1.8 基因检测
WFS1基因外显子8中有1个突变,c.975C>A,p.N325K(编码区第975号核苷酸由胞嘧啶变异为腺嘌呤,导致第325号氨基酸由天冬氨酸酰胺变异为赖氨酸),为杂合错义突变。在gnomAD数据库中未见频率报道。经SIFT软件预测该突变为Affect protein function,经Polyphen-2软件预测该突变为Probably damaging。美国医学遗传学与基因组学学会(ACMG)证据显示该位点临床意义未明。对其姑姑、妹妹、舅舅及姨妈进行的家系验证发现其舅舅及姨妈与患者突变位点一致。
1.9 诊断
WFS1基因相关的不完全型WS。
2 诊疗过程
患者住院期间予以左卡尼汀、多奈哌齐、左乙拉西坦、维生素B1、B12、维生素E及降糖药物等治疗。住院期间头痛发作一次,为额顶部胀痛,不剧,持续数小时自行缓解,自述言语困难及记忆力下降较前变化不明显。出院后1个月电话随访,患者病情稳定,无头痛发作。
3 讨论
WS是一种主要以青少年起病的糖尿病、糖尿病尿崩症、视神经萎缩、耳聋和神经变性为临床特征的常染色体隐性遗传病。首次是于1938年由Wolfram和Wagener对8名兄弟姐妹中有4名患有糖尿病和视神经萎缩的家系进行报道
[7]。1995年,Barrett,Bundey和 Macleod对45例WS的患者的临床特征进行了详细的描述并制定了该病的诊断标准
[1]。根据国际疾病分类草案(ICD-11),WS被分类为一种罕见特殊类型的糖尿病。除上述典型临床表现外,高达60%~90%的患者还存在泌尿系统病变,包括神经源性膀胱、尿失禁和尿路感染等
[8]。其他常见表现有小脑性共济失调、构音障碍、吞咽困难、嗅觉和味觉减退、自主神经功能障碍、头痛、以及焦虑抑郁、惊恐发作等精神症状
[8]。内分泌系统病变可表现为月经初潮推迟、促肾上腺皮质激素及生长激素缺乏、性腺机能不全及低钠血症
[9,10]。WS目前已知的致病基因有两个:WFS1和WFS2
[11,12]。其中90%以上的突变位点位于WFS1。WFS1基因编码的Wolframin蛋白定位于内质网(ER),在脑、胰岛β细胞、肺、胎盘和心脏中高表达
[13],其参与内质网稳态维持
[14]。WFS1异常可引起ER应激,使细胞丧失功能和凋亡
[13]。因此目前认为WS是一种ER功能障碍相关疾病。
本例患者急性起病,头痛后出现言语障碍及记忆力下降,定位于左侧颞叶、额叶及海马,同时存在青年起病的糖尿病、耳聋、双眼高度近视且OCT检查提示神经纤维层厚度局限变薄以及先兆偏头痛,全外显子基因检测发现WFS1基因位点突变经软件预测可影响蛋白功能且很有可能有害,与患者临床表型较为吻合,另外结合家族史,考虑患者为常染色体显性遗传的不完全型WS。
我们首次报道了位于WFS1基因8号外显子区域的突变位点c.975C>A,该位点突变所导致的临床表型与既往报道中多数患者因糖尿病和视神经萎缩症状就诊有所不同。本例患者首诊因突出的神经系统功能障碍而就诊。该患者既往有先兆偏头痛病史,此次病程初期有偏头痛,这与文献报道一致
[15],头痛的具体原因不明确,可能与自主神经功能障碍或神经病有关
[8]。WS的神经系统表现常见的有共济失调、认知障碍、中枢性呼吸衰竭、周围神经病、自主神经病、癫痫、焦虑抑郁及精神症状
[6,15⇓⇓-18],其它不常见症状包括肌阵挛、偏头痛、嗅觉和味觉减退及帕金森综合征等
[1,15,19]。颅脑MRI最常见的表现为脑干、小脑萎缩、视神经、视交叉、视束萎缩及垂体后部TI高信号缺失
[6,15,16,19,20],其他还有脑室周围白质改变、尾状核萎缩、胼胝体肿胀、脑室扩大、脑积水等
[15,17,19,21,22],脑梗死较少见
[1,23]。本例患者多次颅脑MRI检查均发现左侧颞叶、岛叶DWI高信号,在首诊医院被疑诊为自身免疫性脑炎,完善血清及脑脊液自身免疫性脑炎抗体谱为阴性,不支持该诊断。本例患者急性起病,既往也有以急性脑梗死起病的WS
[23],但因本例患者影像学表现不符合血管分布,故不考虑急性脑梗死。ANA谱+ANCA、抗心磷脂抗体、甲状腺功能、TPPA、RPR、维生素B12、叶酸、乳酸、肝肾功等均无异常,因此无免疫、感染、代谢病因相关证据。本例患者的左侧颞叶及岛叶受累考虑与WFS1基因突变所致内质网功能障碍有关。
本例患者心脏超声提示左室心肌致密化不全,这在既往文献中较为罕见。仅在两篇文献中报道过先天性心脏病如法洛四联症、肺动脉瓣狭窄及室间隔缺损等
[2,24]。心肌致密化不全(NVM)是以心室内异常粗大的肌小梁和交错的深隐窝为特征的一种与基因相关的遗传性心肌病
[25]。遗传模式通常为常染色体显性遗传。主要临床表现有心力衰竭、心律失常和体循环栓塞。结合患者家族史(患者母亲死于心力衰竭,舅舅有冠脉支架植入术),考虑患者心脏病变可能与遗传相关。另外,WFS1基因编码的Wolframin蛋白在心脏中高表达,因此可以推测WFS1基因突变导致Wolframin蛋白功能障碍时可能累及心脏,进而出现心脏病变。
WS目前尚缺乏有效的治疗措施,因此早发现、早诊断、早干预对患者至关重要。目前经FDA批准用于ER应激相关疾病的药物有4-苯基丁酸和牛磺熊去氧胆酸
[26-27]。另外,有两种特定针对WS患者的药物丹曲林和丙戊酸钠目前正在进行药物临床试验(ClinicalTrials.gov: NCT02829268, NCT03717909)
[3,28,29]。WS作为多系统受累及的综合征,需通过多学科合作包括内分泌科、神经科、眼科、耳鼻喉科、泌尿科及心内科等开展早期干预、综合防控,从而延缓其进展。
总之,在临床实践中,对于有多系统累及的患者,除考虑免疫、代谢、肿瘤等相关病因外,相对罕见的遗传性疾病也应纳入鉴别诊断,以免误诊。对于WS的文献复习总结见表1。
表1 Wolfram综合征患者的临床表现及颅脑MRI等临床资料汇总Tab 1 clinical characteristics of patients with Wolfram syndrome |
| 文献 | 年份 | 病例数 | 年龄
(岁) | 性别
(男/女) | 临床表现 | 脑MRI |
|---|
| Morgia等[14] | 2020 | 10 | 16-56 | - | DIDMOAD,偏头痛,早熟,
中枢性睡眠呼吸暂停,吞咽困难,高血压,青光眼,痴呆,
共济失调,甲状腺功能减退 | 视神经通路前部萎缩(10/10)
TI神经垂体“亮信号”(10/10)
脑桥萎缩(10/10)
脑桥T1低信号(10/10)
脑白质T2高信号(5/10)
第三脑室扩大(4/10)
小脑萎缩(4/10)
延髓萎缩(4/10)
顶枕叶萎缩(3/10)
中脑萎缩(2/10) |
| Licis等[19] | 2019 | 15 | 8.9-29.7 | 6/9 | DIDMOAD,中枢性睡眠呼吸暂停 | 脑桥、延髓、小脑萎缩 |
| Rove等[15] | 2018 | 36 | 7-30 | 13/23 | DIDMOAD,尿路症状 | 脑桥萎缩 |
| Gowda等[30] | 2018 | 1 | 23 | 女 | DIDMOAD,肾积水,共济失调,
认知下降,精神分裂症 | 脑桥、小脑萎缩 |
| Harsha等[31] | 2016 | 1 | 27 | 男 | 糖尿病、耳聋、进行性视力下降,性腺机能减退 | 脑桥腹侧T2高信号,垂体后部T1高
信号,双侧视神经萎缩 |
| Lugar等[32] | 2016 | 21 | 14.33±5.8 | 8/13 | DIDMOAD,共济失调,认知下降 | 脑桥腹侧、小脑白质、视觉皮层、
白质纤维束萎缩 |
| Zmyslowska等[16] | 2014 | 7 | 10.1-16.0 | 0/7 | DIDMOAD,共济失调,多发性神经病眼震,腱反射减低或消失,
吞咽困难,认知下降 | 视神经、视交叉、视束萎缩脑干萎缩
,侧脑室旁白质病变 |
| Vale等[33] | 2013 | 1 | 32 | 男 | DIDMOAD,低钠血症 | 视神经、脑桥、小脑萎缩 |
| Lieber等[17] | 2012 | 1 | 61 | 男 | 糖尿病,自主神经病,视神经萎缩,
认知下降,抑郁,共济失调 | 大脑半球、小脑、脑干萎缩,脑白质
改变 |
| Hershey等[34] | 2012 | 11 | 5.9-25.8 | 5/6 | DIDMOAD,焦虑,抑郁,共济失调 | 脑干、小脑萎缩 |
| Waschbisch等[18] | 2011 | 1 | 44 | 女 | DIDMOAD,中枢性睡眠呼吸暂停,吞咽困难,认知下降,眼震,
肌阵挛,嗅觉减退,抑郁,共济失调,帕金森综合征 | 脑干、小脑、胼胝体、胼胝体、尾状核萎缩,视神经、视交叉萎缩,脑室旁T2高信号 |
| Kesavadev等[35] | 2011 | 1 | 31 | 女 | 糖尿病,视神经萎缩,白内障,尿崩症,一过性精神症状,抑郁 | 全脑萎缩(皮层、脑干、小脑) |
| Ganie等[36] | 2011 | 7 | 17.5-7.34 | 5/2 | 糖尿病、视神经萎缩、耳聋(4/7)、
尿崩症(6/7)、痉挛性肌阵挛、胰腺
性吸收不良伴身材矮小、紫绀性心脏病、黄疸和胆管炎 | 脑干萎缩(4/7),垂体后叶TI高信号缺失(6/7),空蝶鞍(1/7) |
| Chaussenot等[5] | 2011 | 59 | 5-55 | 32/27 | 糖尿病(57/59)、视神经萎缩
(58/59)、尿路症状(31/59)、神经系统并发症(共济失调、
周围神经病、认知下降、癫痫、精神发育迟滞等)(321/59)、
耳聋(27/59)、尿崩症(17/59)、精神症状(23/59)、
胃肠道症状(7/59)、白内障(3/59)、性腺萎缩(8/32) | 脑桥、小脑、顶枕叶、视神经、视交叉萎缩,神经垂体T1高信号缺失,白质脑病,脑室旁高信号,额叶皮质沟回T2异常信号 |
| Synofzik等[37] | 2010 | 1 | 39 | 男 | 糖尿病、视神经萎缩、直立性低血压、晕厥、认知下降、淡漠、
退缩、尿失禁、眼震、共济失调 、肌张力障碍 | 脑桥、小脑萎缩 |
| Labauge等[38] | 2010 | 1 | 29 | 女 | 糖尿病、视神经萎缩、耳聋、共济失调、抑郁、认知下降、周围
神经病 | 全脑萎缩(脑干著)、弥漫性白质脑
病,侧脑室后角明显 |
| Hong等[39] | 2009 | 1 | 13 | 女 | DIDMOAD | 视神经萎缩、视辐射异常信号、垂体
后叶TI高信号缺失 |
| Jockschat等[40] | 2009 | 1 | 21 | 男 | DIDMOAD、攻击性行为、冲动控制障碍、自杀、认知下降 | 视神经萎缩、黑质高信号、垂体
后叶TI高信号缺失 |
| Ito等[41] | 2007 | 1 | 35 | 男 | DIDMOAD | 脑干和小脑中脚萎缩,小脑轻度萎缩、垂体后叶T1高信号缺失,视神经萎缩 |
| Pakdemirli等[42] | 2005 | 1 | 32 | 女 | DIDMOAD、构音障碍、认知下降、焦虑抑郁 | 脑干和小脑蚓部显著萎缩,小脑半球
中度萎缩、垂体后叶T1高信号缺失,
视神经、视交叉、视束萎缩,侧脑室后角 白质高信号 |
| Inukai等[43] | 2005 | 2 | 29,41 | 1/1 | DIDMOAD,亚临床垂体功能减退 | 脑桥萎缩 |
| Hadidy等[21] | 2004 | 16 | 男:10-37
女:7-35 | 6/10 | - | 脑干、小脑和视通路萎缩,前庭蜗神经核T2高信号,垂体后叶高信号消失 |
| Simsek等[44] | 2003 | 9 | 6-18 | 6/3 | DIDMOAD,生长发育迟缓,初潮推迟,性腺功能减退,尿路症状 | 脑干、小脑、皮层、下丘脑萎缩、
右侧颞叶梗死、垂体后叶T1高信号缺失 |
| Shannon等[45] | 1999 | 1 | 32 | 女 | DIDMOAD,认知下降,感觉异常,踝反
射消失,双足震动觉减退,共济失调 | 脑干、小脑、皮层萎缩,视辐射、
脑室旁、胼胝体T1高信号 |
| Galluzzi等[46] | 1999 | 1 | 12 | 女 | DIDMOAD,尿路症状,多发性感觉 运动神经病,腱反射消失 | 垂体后叶T1高信号缺失,视神经、视
交叉、视束萎缩,黑质T2高信号,下丘脑发育不良 |
| Barrett等[1] | 1995 | 45 | 6-30 | 21/24 | DIDMOAD,尿路症状,躯干/步态共济失调、肢体反射减弱,
肌阵挛,水平性眼震,中枢性呼吸暂停,小脑性构音障碍,
自主神经病,味觉嗅觉消失,偏瘫,触觉减退 | 全脑萎缩(脑桥、延髓 、小脑显著),垂体后叶T1高信号缺失,视神经信号减少 |
| Rando等[47] | 1992 | 2 | 19,21 | 女 | 糖尿病、尿崩症、智力迟钝、步态不稳、视力下降、尿路症状、
阅读障碍、暴食症、抑郁症、边缘人格障碍 | 脑干、小脑、皮层萎缩、视神经萎缩、垂体后叶T1高信号缺失 |


{kind=link}
{kind=link}